
Molecular differences between HLA alleles vary up to 57 nucleotides
within the peptide binding coding region of human Major
Histocompatibility Complex (MHC) genes, but it is still unclear whether
this variation results from a stochastic process or from selective
constraints related to functional differences among HLA molecules.
Although HLA alleles are generally treated as equidistant molecular
units in population genetic studies, DNA sequence diversity among
populations is also crucial to interpret the observed HLA polymorphism (
2011
HLA DNA Sequence Variation among Human Populations: Molecular Signatures of Demographic and Selective Events.
PLoS ONE 6(2):
e14643.
doi:10.1371/journal.pone.0014643).
Traditionally, serology has been used for HLA typing for many decades; however, serological typing of histocompatibility class II molecules depends on the adequate expression of these molecules on the surface of B lymphocytes, the availability of viable cells and a complete set of antisera. However, the application of molecular methods (RFLP, PCR, SSO etc.) to HLA typing has led to the situation whereby nearly every laboratory performs some DNA typing for the detection of HLA alleles.
Why HLA loci are so important?
HLA loci present the highest degree of polymorphism of all human genetic
systems. Prior knowledge of the extent of diversity is essential in the
development and selection of molecular typing methods. Reliable allele
frequencies are also important in allogeneic unrelated hematopoietic
stem cell transplantation to determine the likelihood of finding
closely HLA matched donors for each patient.The genetic diversity of the HLA loci is responsible for the efficiency
of the immune system in eliminating cells carrying foreign antigens.
There is a need to develop a measure of this genetic diversity in order
to assess how different populations are equipped to respond to
foreign-antigen exposure, and to evaluate the contribution of each HLA
locus.
The HLA system has been extensively studied from an evolutionary
perspective. The region contains a number of closely linked genes whose
products control a variety of functions concerned with the regulation
of immune responses. In addition, the genetic predisposition to over 40
diseases maps to this region. A number of observations indicate that
strong selection is acting on the HLA region, including its extensive
polymorphism with very even allele frequencies, the preferential
occurrence of high levels of variability at positions critical to
antigen recognition, the great age of alleles and the patterns of
linkage disequilibrium among loci. The form of the selection is
unknown. Although balancing selection is a strong candidate, it seems
unlikely that only one selective mechanism is operating in this complex
multigene family region. Mutation, recombination and gene conversion
all contribute to the generation of HLA variability. The apparent great
age of many HLA alleles revealed by phylogenetic analysis suggests that
the absolute rate of production of new variants is not high. Detailed
studies of population and evolutionary features of the HLA region are
necessary for an informed discussion of the evolution of disease
predisposing genes and epitopes, and of complex multigene families (Thomson G.HLA population genetics.1991 Jun;5(2):247-60.)
Each HLA allele name has a unique number corresponding to up to four sets of digits separated by colons. The length of the allele designation is dependant on the sequence of the allele and that of its nearest relative. All alleles receive at least a four digit name, which corresponds to the first two sets of digits, longer names are only assigned when necessary.The digits before the first colon describe the type, which often corresponds to the serological antigen carried by an allotype. The next set of digits are used to list the subtypes, numbers being assigned in the order in which DNA sequences have been determined. Alleles whose numbers differ in the two sets of digits must differ in one or more nucleotide substitutions that change the amino acid sequence of the encoded protein. Alleles that differ only by synonymous nucleotide substitutions (also called silent or non-coding substitutions) within the coding sequence are distinguished by the use of the third set of digits. Alleles that only differ by sequence polymorphisms in the introns or in the 5' or 3' untranslated regions that flank the exons and introns are distinguished by the use of the fourth set of digits (further information: http://hla.alleles.org/nomenclature/naming.html).
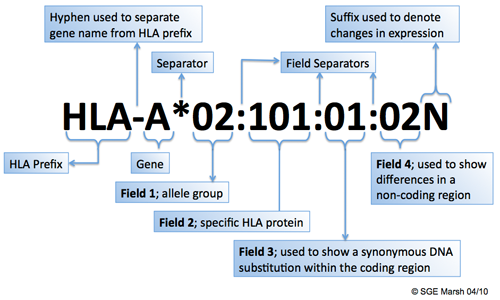
Example
HLA-A identifies HLA A locus
HLA-A1 serologically defined antigen
HLA-A* asterisk denotes HLA alleles defined by molecular methods
HLA-A*01 2 digit resolution denotes a group of alleles corresponds usually to serological group – low resolution
HLA-A*0101 4 digit resolution – sequence variation between alleles results in amino acid substitutions
HLA-A010101 6 digit resolution – non coding variation: sequence changes synonymous no amino acid substitution
HLA types and linked SNPs
Still, classical HLA alleles (e.g. HLA-, HLA-B, etc.) are tricky to analyze
with chip technology employed in popular commercial personal genomic
services (23andme, FTDNA's Family Finder and deCODEme) , because analysis is a complex process, requiring a large number of multiplex PCR reactions to obtain a patient's full genotype. That is why classical HLA typing methods are often imparctical
for large-scale studies.
Fortunately for us , Wellcome Trust Centre for Human Genetics described a method for imputing classical alleles from linked SNP genotype data, implemented in the imputation framework (HLA*IMP) (Alexander Dilthey, Loukas Moutsianas, Stephen Leslie, and Gil McVean. HLA*IMP – an integrated framework for imputing classical HLA alleles from SNP genotypes Bioinformatics btr061 first published online February 7, 2011 doi:10.1093/bioinformatics/btr061).
HLA*IMP imputes HLA type information based on SNP genotype, using methods of iterative model building to select a set of informative SNPs for particular supported genotyping chips (AffyMetrix 500K, AffyMetrix 900K, Illumina 300K, Illumina 550K,Illumina 650K, Illumina 1M). Thus, HLA*IMP enables researchers to perform classical HLA allele imputation from genotype data collected from several available genome-wide SNP sets through reference to a reference data set of over 2,500 samples of European ancestry with dense SNP data and classical HLA allele types. This reference set includes:
- The 1958 Birth Cohort typed both on the Illumina 1.2M and Affymetrix Genome-Wide Human SNP Array 6.0 chips (TheWellcome Trust Case Control Consortium, 2007) - 2420 genotype samples x 7733 SNP in the extended HLA region.
- The HapMap CEU samples (The International HapMap Consortium, 2007) and CEPH CEU+ additional samples (de Bakker et al., 2006) - 92 samples x 7733 BC58-overlapping SNPs)
SNP haplotypes for the 1958BC and CEU+ samples were phased using IMPUTE v2 using the trio-phased HapMap samples as a reference dataset. Classically typed HLA genotypes were then phased into SNP haplotypes by using PHASE (Stephens and Scheet, 2005) applying standard settings for multiallelic loci. The combined reference dataset consists of 5024 haplotypes with data on 7733 SNPs in the HLA region. This splits up into 2474 (HLA-A), 3090 (HLA-B), 2022 (HLA-C), 175 (HLA-DQA1), 2629 (HLA-DQB1), 2665 (HLA-DRB1) locus-specific haplotypes which are used for inference.
The experiment with MDLP dataset
Motivation:
As i previously mentioned in my blog (re: Chromosome 6), 23andme's RelativeFinder and AncestryFinder revealed (among my results) a cluster of a
prominent number of HIR-matches (315), positioned in the exactly the
same subregion of HLA-MHC domain on Chr.6 (21Mb-38Mb). This remarkable
group of matches constitutes almost a half of my total AF/RF matches
(315/720 or 43,75%).
Earlier i suggested that such a preponderance of HIR-matches to HLA region is indicative of case of sharing the same extended HLA haplotype. Until recently, my suggestion rested solely on my intiution. Then, i found solution to HLA*IMP challenges and so far, i've managed to make HLA*IMP work with 23andme genotyper (Illumina Omnio Express) call raw data.
Earlier i suggested that such a preponderance of HIR-matches to HLA region is indicative of case of sharing the same extended HLA haplotype. Until recently, my suggestion rested solely on my intiution. Then, i found solution to HLA*IMP challenges and so far, i've managed to make HLA*IMP work with 23andme genotyper (Illumina Omnio Express) call raw data.
For my test, i needed to make sure that my data meets the following requirements:
- SNPs must be from the xMHC region (chromosome 6)
- European ancestry of the typed individuals
- sufficient quality and typed SNP density in the HLA region to enable reliable phasing
- since HLA*IMP doesn't provide a direct support for 23andme's chips and i was using the combined set of genotypes from 23andme's chipset v2 and chipset v3, i've choosed "a downgraded" version of Illumina genotyping platform (Illumina 300K)
In order to my initial assumption of HLA haplotype sharing i selected 7 participants from my project (including 4 individuals without HIR-sharing on Chromosome 6 (not detected by 23andme's AF) me, my mother and an individual, who has a HIR-match with me and my mother in xMHC region). I converted 23andme's raw data of the project's participants into Plink format, merged the files into one dataset and extracted SNPs on Chromosome 6 using Plink command --chr 6. After that, i converted the genotype data from Plink format to HLA*IMP input data format. As a next step, i carried out quality control on the genotype data, removing SNPs and individuals with too much missing data and aligning complementary SNPs to the HapMap reference strand. Finally, i phased included genotypes to obtain haplotype data. Note: each individual was anonymized by substituting project's ID with prefix N.
The haplotype data was uploaded to HLA*IMP back-end web service for imputing HLA types.
The imputed HLA types look as follows (each individual is represented by 2 HLA haplotypes in HLA-A:HLA-B:HLA-C:HLA-DQA:HLA-DQB:HLA-DRB format)
| IndividualID | Chromosome | HLAA | HLAB | HLAC | HLADQA | HLADQB | HLADRB | ||
| N1 | 1 | 101 | 801 | 701 | 501 | 201 | 301 | ||
| N1 | 2 | 2601 | 2705 | 102 | 101 | 501 | 101 | ||
| N6 | 1 | 3101 | 801 | 701 | 501 | 201 | 301 | ||
| N6 | 2 | 201 | 1501 | 304 | 501 | 201 | 301 | ||
| N3 | 1 | 6801 | 1501 | 102 | 101 | 501 | 101 | ||
| N3 | 2 | 2301 | 5201 | 501 | 101 | 501 | 101 | ||
| N2 | 1 | 101 | 801 | 701 | 501 | 201 | 301 | ||
| N2 | 2 | 2601 | 3801 | 1203 | 102 | 602 | 1501 | ||
| N5 | 1 | 301 | 1501 | 304 | 501 | 302 | 401 | ||
| N5 | 2 | 205 | 5001 | 602 | 501 | 202 | 701 | ||
| N7 | 1 | 101 | 801 | 701 | 501 | 301 | 1101 | ||
| N7 | 2 | 101 | 1501 | 303 | 103 | 604 | 1301 | ||
| N4 | 1 | 301 | 702 | 702 | 401 | 402 | 801 | ||
| N4 | 2 | 2402 | 4002 | 202 | 501 | 301 | 1101 |
The haplotypes should be read as follows (for example, in case of N1): HLA A*0101 : Cw*0701 : B*0801 : DRB1*0301 : DQA1*0501 : DQB1*0201.
From the table posted above, one can easily mention a matching pattern between N1, N2 and N7, as they share the similiar haplotype. As a matter of fact N1 (ny mother), N2 (me) and N7 share HIR-segment (matching DNA segment equal to or greater than seven Centimorgans (abbreviated as cMs) on one segment with 700 or more SNPs), found by RelativeFinder service at 23andme.
Thus, it is safe to conclude that the imputation was accurate and my initial suggestion seems to be backed up by HLA imputation
The practical implications of test
Aside from the medical usefulness, there are also some benefits of knowing HLA type for genetic genealogists:
1) First of all, it is possible to determine the nature of the sharing the segments in xMHC region on Chromosome 6. Consider the aforementioned extended haplotype HLA A*0101 : Cw*0701 : B*0801 : DRB1*0301 : DQA1*0501 : DQB1*0201 (in shorthand: A1::DQ2).A1::DQ2 haplotype creates a conundrum for the evolutionary study of recombination.A1::DQ2 does not follow the expected dynamics. Other haplotypes exist in the region of Europe where this haplotype formed and expanded, some of these haplotypes also are ancestral and also are quite large. At 4.7 million nucleotides in length and ~300 genes the locus had resisted the effects of recombination, either as a consequence of recombination-obstruction within the DNA, as a consequence of repeated selection for the entire haplotype, or both.
The length of the haplotype is remarkable because of the rapid rate of
evolution at the HLA locus should degrade such long haplotypes. A1::DQ2 is the most frequent haplotype of its length found in US Caucasians, ~15% carry this common haplotype. The SNP analysis of the haplotype suggests a potential founding affect of 20,000 years within Europe, though conflicts in interpreting this information are now apparent. The last possible point of a constriction forcing climate was the Younger Dryas before 11,500 calendar years ago, and so the haplotype has taken on various forms of the name, Ancestral European Haplotype, lately called Ancestral Haplotype A1-B8 (AH8.1). It is one of 4 that appear common to western Europeans and other Asians. Assuming that the haplotype frequency was 50% at the Younger Dryas and declined by 50% every 500 years the haplotypes should only be present below 0.1% in any European population. Therefore it exceeds the expected frequency for a founding haplotype by almost 100 fold. For genetic genealogist, the said would mean that the hotspot of DNA matching in xMHC region could be indicative of very distant common ancestor, probably as recent as Neolithic era.
2)It is also possible to infer the geographic ditribution of a particular HLA haplotype. Using the same A 1::DQ2 haplotype, we could see that it is found in
Iceland, Pomors of Northern Russia, the Serbians of Northern Slavic
descent, Basque, and areas of Mexico where Basque settled in larger
numbers. The haplotypes great abundance in the most isolated geographic
region of Western Europe, Ireland, in Scandinavians and Swiss suggests
that low abundance in France and Latinized Iberia are the result of
displacements that took place after the Neolithic onset. This implies a
founding presence in Europe that exceeds 8000 years.
The database of allele frequencies is a very convenient tool for analyzing HLA haplotype frequency in a worldwide scale.
| 1 | A*01:01-B*08:01-C*07:01-DRB1*03:01-DQB1*02:01 |  | Ireland South |
11.50
|
250
| ||
| 2 | A*01:01-B*08:01-C*07:01-DRB1*03:01:01-DQB1*02:01 |  | England North West |
9.50
|
298
| ||
| 3 | A*01:01-B*08:01-C*07:01-DRB1*03:01-DQB1*02:01-DPB1*04:01 | | Ireland South |
8.30
|
250
| ||
| 4 | A*01:01-B*08:01-C*07:01-DRB1*03:01-DQB1*02:01 |  | Poland |
4.00
|
200
| ||
| 5 | A*01:01-B*08:01-C*07:01-DRB1*03:01-DQB1*02:01 |  | USA Hispanic pop 2 |
1.78
|
1,999
| ||
| 6 | A*01:01-B*08:01-C*07:01-DRB1*03:01-DQB1*02:01-DPB1*01:01 | | Ireland South |
1.40
|
250
| ||
| 7 | A*01:01-B*08:01-C*07:01-DRB1*03:01-DQB1*02:01 | | USA African American pop 4 |
1.39
|
2,411
| ||
| 8 | A*01:01-B*08:01-C*07:01-DRB1*03:01-DQB1*02:01 | | USA Asian pop 2 |
0.09
|
1,772
|

Good blogpost.
ReplyDeleteHello,
ReplyDeleteWhat a post :) Great work. I will use your ideas in future. Thanks for sharing. Keep it up !!! Visit college paper for best papers.
Your natural talent and kindness in handling all the details was excellent. I don't know what I would've done if I hadn't come upon such a thing like this. Visit is raeli jewellery for best Jewellery.
ReplyDelete